https://www.gosh.nhs.uk/conditions-and-treatments/clinical-outcomes/cystic-fibrosis-clinical-outcomes/
Cystic fibrosis clinical outcomes
Clinical outcomes are measurable changes in health, function or quality of life that result from our care. Constant review of our clinical outcomes establishes standards against which to continuously improve all aspects of our practice.
About the Cystic Fibrosis Service
Cystic fibrosis (CF) is a life-limiting disorder that affects more than 10,000 people in the UK. A little under half of those with the condition are children. CF is caused by a faulty gene that causes the body to produce abnormally thick and sticky mucus. It is a multisystem disorder, involving many internal organs but primarily affecting the lungs and digestive system.
The Cystic Fibrosis Unit at Great Ormond Street Hospital (GOSH) is a designated specialist centre, which cares for approximately 200 children from newborn to 16 years of age. Most referrals come from the National Cystic Fibrosis Newborn Screening Programme.
We have a full specialist multidisciplinary team of doctors, nurses, physiotherapists, dietitians, pharmacists, psychologists, social workers and administrative staff. The unit works closely with a number of other departments within GOSH, in particular paediatric radiology, endocrinology and gastroenterology. In a small proportion of cases, the care of children is shared between a local ‘network’ hospital and the specialist service at GOSH. Where this arrangement takes place, the outreach team provides a link between the centre and local team.
There is a strong research collaboration between GOSH and the other London cystic fibrosis specialist paediatric units. We are part of the CTAP (Clinical Trials Accelerator Platform), a research programme that engages pharmaceutical companies and researchers to invest in CF clinical trials. This has increased the number of clinical trial opportunities, and improved access to the newest therapies for our CF patients. The department also has an international reputation for research, particularly in respiratory physiology and measuring lung function in babies.
Clinical outcome measures
1. Lung function
2. Nutritional status
3. Pseudomonas aeruginosa infection rate
How to interpret the graphs
There is natural variation between centres because of differences between the patients receiving care. Using only the national average as a standard makes it difficult to tell if a centres’ outcome are within ‘expected’ variation. For this reason, the funnel plots also show ‘control limits’: the dashed curved lines on the graphs that give them the ‘funnel’ shape. Each graph shows the national average result for all paediatric CF centres in the UK as a horizontal red line in the middle of the funnel.
If the result for a CF centre is between the two control limits its results are expected. If a result is below the bottom control it is lower than expected, if it is above the upper control limit it is higher than expected.
Key to the graphs:

GOSH's clinic ID is 90. The full list of clinics and their IDs can be found on page 76 of the UK Cystic Fibrosis Registry Annual Data Report 2024 on the Cystic Fibrosis Trust website.
1. Lung function, measured by: Forced Expiratory Volume in one second (FEV1)
One of the key measures undertaken to monitor CF lung function is the Forced Expiratory Volume in one second (FEV1). This measures the volume of expired air blown out in the first second during a hard and fast breath out. This measure is then compared to that of an average child of the same age, sex, height and ethnicity, who does not have CF. This comparison is against ‘population norms’ to provide a benchmark for a normal range.
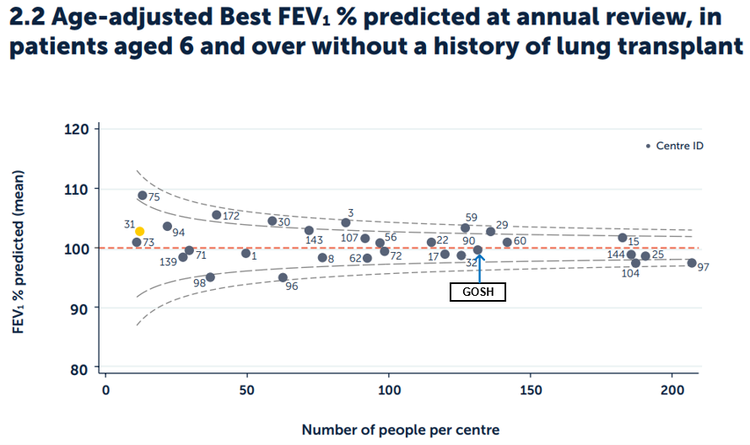
Figure 2.2. Age-adjusted Best FEV1% predicted in patients aged six and over without a history of lung transplant, by paediatric centre/clinic.
Published with the permission of the UK Cystic Fibrosis Registry
In 2024, the national average FEV1 % predicted for all those with CF aged 6 and over was 100%. The result for our clinic at GOSH in 2024 was 99.6%, within the control limits of expected results. It is our goal to transfer our young people to adult services with normal lung function.
2. Nutritional status, measured by: Body Mass Index (BMI)
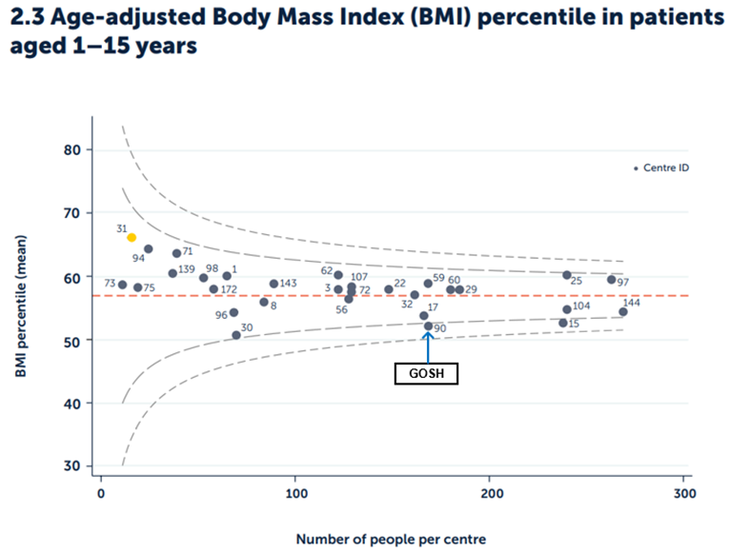
Body mass index (BMI) is a measure of nutritional status.
It is a number calculated from weight and height. BMI shows weight in relation to height. In CF, a healthy BMI has been shown to be associated with better outcomes including lung function. In children, we report BMI percentiles (comparing the child’s BMI within the range of children of the same sex and age who do not have CF). This comparison is against population norms to provide a benchmark for a normal range. This is reported at one timepoint only (annual assessment).
Figure 2.3 Age adjusted BMI percentile among patients aged one to 15 years by paediatric centre or clinic.
Published with the permission of the UK Cystic Fibrosis Registry
In 2024, the national average BMI percentile for all children with CF aged one to 15 was 57%. The BMI percentile for our clinic at GOSH in 2024 was 52.2%, similar to the previous year.
Evidence is emerging that there is no additional clinical benefit to having a high BMI in CF. National guidelines for CF recommend a BMI percentile of 50 or greater is achieved and maintained. However, a BMI in the range of 25-75th percentile is regarded as a healthy BMI in all children, and the current GOSH BMI percentile of 52.2% is within this range.
BMI percentile in isolation should be interpreted with caution. In clinic it is discussed in conjunction with weight and height percentiles, lung function and clinical status. BMI does not reflect body composition (fat mass and lean mass). Lean mass is more strongly associated with lung function than BMI. Weight, height, dietary intake and absorption of children with CF are continually assessed to ensure steady weight gain and growth is achieved. The nutritional status of each individual patient is reviewed as a priority by the CF Dietetic Team to ensure timely nutrition interventions are recommended. Interventions include optimising dietary absorption using enzymes, high calorie diets, oral nutrition supplements and the recommendation of enteral tube feeding when appropriate.
3. Pseudomonas aeruginosa infection rate
The lungs and airways are one of the primary systems affected by CF, so it is essential that we do as much as we can to prevent and actively treat chest infections to avoid lung damage. Pseudomonas aeruginosa is a bacterium that can cause infection in people with CF. Early aggressive treatment and careful attention to prevention of cross-infection aims to reduce the number of children with chronic Pseudomonas aeruginosa infection.
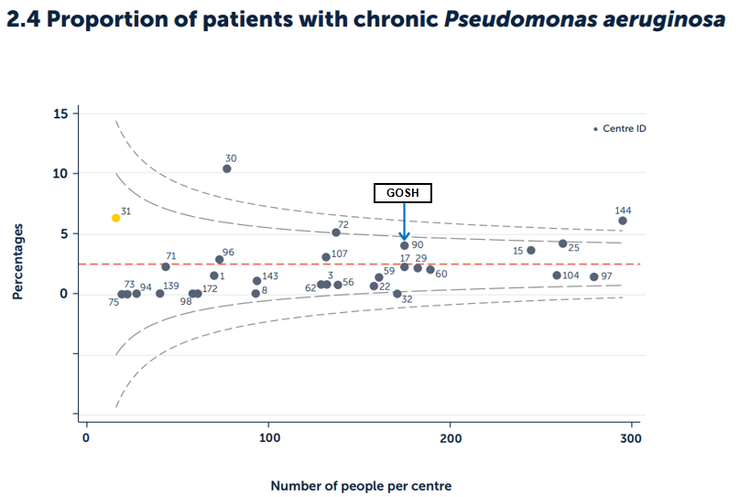
Figure 2.4 Proportion of patients with chronic Pseudomonas aeruginosa by paediatric centre or clinic.
Published with the permission of the UK Cystic Fibrosis Registry
In 2024, the national average percentage of patients with chronic Pseudomonas aeruginosa infection in paediatric centres in the UK was 2.5%. The proportion of GOSH patients with chronic Pseudomonas aeruginosa was 4%, which is within the control limits.
For this outcome measure, a lower % figure is more favourable. Although we continue to be reassured by this data, we certainly cannot be complacent and close attention to cross infection remains high on our list of priorities.
Continuous improvement
Our annual clinic surveillance programme for pseudomonas cross infection continues, where we take samples from as many children as possible in a two-month period and any that grow pseudomonas are sent to a specialist laboratory for ‘typing’ to alert us to any cross-infection potential or particularly virulent strains. To date, there has been no evidence of either within the GOSH clinic.