https://www.gosh.nhs.uk/conditions-and-treatments/clinical-outcomes/metabolic-medicine-clinical-outcomes/
Metabolic Medicine clinical outcomes
Clinical outcomes are measurable changes in health, function or quality of life that result from our care. Constant review of our clinical outcomes establishes standards against which to continuously improve all aspects of our practice.
About the Metabolic Medicine Service
Great Ormond Street Hospital’s (GOSH) Metabolic Medicine Service is the largest paediatric metabolic centre in the UK. We are the principal provider of specialist children’s metabolic medicine services for London (north of the Thames) and the surrounding counties. We provide a national and international service for the management of rare and complex paediatric metabolic conditions.
We see patients from 0 to 16 years old with suspected or diagnosed metabolic disease, including disorders of intermediary metabolism, neurometabolic disorders, congenital disorders of glycosylation (CDG), disorders detected on newborn screening, galactosaemia, familial hypercholesterolaemia (FH) and mitochondrial disorders. We are one of three nationally commissioned NHS specialised services for the management of children with lysosomal storage disorders (LSD).
The Clinical Biochemistry department at GOSH is one of the UK newborn screening centres. The Metabolic Medicine Service provides care for all neonates from our catchment area with positive neonatal screening results, which include phenylketonuria (PKU), medium-chain acyl-CoA dehydrogenase deficiency (MCADD), glutaric aciduria type 1 (GA1), isovaleric acidaemia (IVA), maple syrup urine disease (MSUD), Hereditary Tyrosinaemia Type 1 (HT1) and homocystinuria (HCU).
Our multi-disciplinary metabolic team is comprised of consultants and clinical nurse specialists, with specialist input from metabolic dieticians, biomedical scientists, physiotherapists, clinical psychologists, social workers, and speech and language therapists.
Clinical outcome measures
1. Home treatment for Lysosomal Storage Disorder (LSD)
LSD is an umbrella term for a group of more than 70 genetic disorders that are caused by a lysosomal enzyme deficiency. Worldwide, these conditions affect an estimated one in 7,700 people.
Many of the LSDs display a progressive disease pattern, which means that symptoms get worse over time if left untreated. Treatments for some of the LSDs include enzyme replacement therapy (ERT), which is administered intravenously every week or two, depending on the type of disorder.
It is important for the wellbeing of the child and their family to seek, where possible, to reduce disruption caused by the treatment frequency. Home infusion can help, with fewer days of school missed, less disruption for parents and siblings, and reduced travel and financial implications.
Home infusion can only be an option if it can be established safely with patient/family training, with protocols in case of infusion reactions, and with support from the specialist centre.
1.1 Children with an LSD on home ERT
|
Number of patients with an LSD on home ERT |
2022/23 | 2023/24 | 2024/25 |
|---|---|---|---|
| Number of current patients with an LSD* | 298 | 328 | 346 |
| Number of current patients with an LSD on home ERT | 82 | 99 | 108 |
*Not all LSDs have an available enzyme replacement therapy. Some are treated with other interventions including haematopoietic stem cell transplant. All patients eligible for ERT are offered this when clinically appropriate.
Of the current GOSH patients (as of March 2024/25), for whom homecare is suitable, 100% are on home ERT.
1.2 Children with an LSD on ERT who are transferred to home infusions within three months of commencement of therapy
|
New patients with an LSD on ERT who are transferred to home infusions |
2022/23 – 2024/25 |
|---|---|
| Patients transferred within three months | 9 |
| Patients transferred after three months | 8 |
| Total number of patients | 17 |
| Percentage of patients transferred within three months | 53% |
In 2024/25, 6 (75%) of children were transferred to home ERT within three months of commencement of therapy.
From April 2022 to March 2025, 9 (53%) children were transferred to home ERT within three months of commencement of therapy. 8 (47%) children were transferred after three months because of clinical reasons.
2. Medium-chain acyl-CoA dehydrogenase deficiency (MCADD) seen for review
Medium-chain acyl-CoA dehydrogenase deficiency (MCADD) is a rare inherited genetic disorder in which there is a block in the metabolism of fat into energy when the body's demand for energy increases and calorie intake is often reduced. This means that when someone with MCADD develops an illness, such as diarrhoea and vomiting or an infection that prevents them from eating, they are at risk of becoming more unwell, and they are also at risk of developing hypoglycaemia.
MCADD is a lifelong condition that is present from birth. It is estimated to affect up to 1 in every 8,000 babies born in the UK and is usually picked up using the newborn blood spot test. Medical complications of the condition can usually be prevented, so early detection and medical management is very important.
2.1 Patients with MCADD seen for review within 24 hours
Newly identified patients with MCADD should be seen for a 'face-to-face review' within 24 hours of receiving information about the newborn screening result. At the review, they are provided with necessary information and an emergency regimen, which is a special feeding plan to provide patients with energy if they are unwell or unable to take their normal nutritional intake.
Numerator: The number of newly identified MCADD Newborn Screening (NBS) patients seen for a 'face-to-face review ' by the metabolic clinical team within 24 hours of receiving the screening report and provided with necessary information and emergency regimen.
Denominator: Total number of new positive diagnoses of MCADD by NBS at the centre.
|
MCADD patients seen for review |
2022/23 | 2023/24 | 2024/25 |
|---|---|---|---|
|
Number of newly identified MCADD NBS patients seen for a 'face-to-face' review by metabolic clinical team within 24 hours |
8 | 9 | 10 |
| Total number of new positive NBS diagnoses of MCADD | 9 | 12 | 10 |
|
Percentage of newly identified MCADD NBS patients seen for a 'face-to-face' review by metabolic clinical team within 24 hours |
89% | 75% | 100% |
This measure is a national measure that is reported to commissioners by MCADD specialised services. Between April 2022 and March 2024, all patients received clinically appropriate care and were provided with the necessary information and an emergency regimen. As per national protocol, we see patients on the next working day after diagnosis. Therefore, a small number of patients who were diagnosed on the weekend were seen on the next working day, rather than ‘within 24 hours’. However, those diagnosed on the weekend have an assessment made on the same day via telephone and urgent review arranged if clinically needed.
3. Review of emergency regimen for children with organic acidaemia conditions
Children and young people with metabolic conditions will often need to follow an emergency regimen diet when they become ill (such as vomiting or diarrhoea) to help prevent deterioration of metabolic function and a possible admission to hospital. This involves drinking a special high glucose drink at specific times until they are able to resume their regular diet. This is particularly important for those with a group of conditions called organic acidaemias.
3.1 Patients with PA or MMA (non-vitamin B12 responsive) provided with an emergency regimen
A measure of our effectiveness that we report to NHS England each year is the number of patients with propionic acidaemia (PA) or methylmalonic acidaemia (MMA) that is non-Vitamin B12 responsive, who have an emergency regimen as per the British Inherited Metabolic Disease Group guidelines (www.bimdg.org.uk).
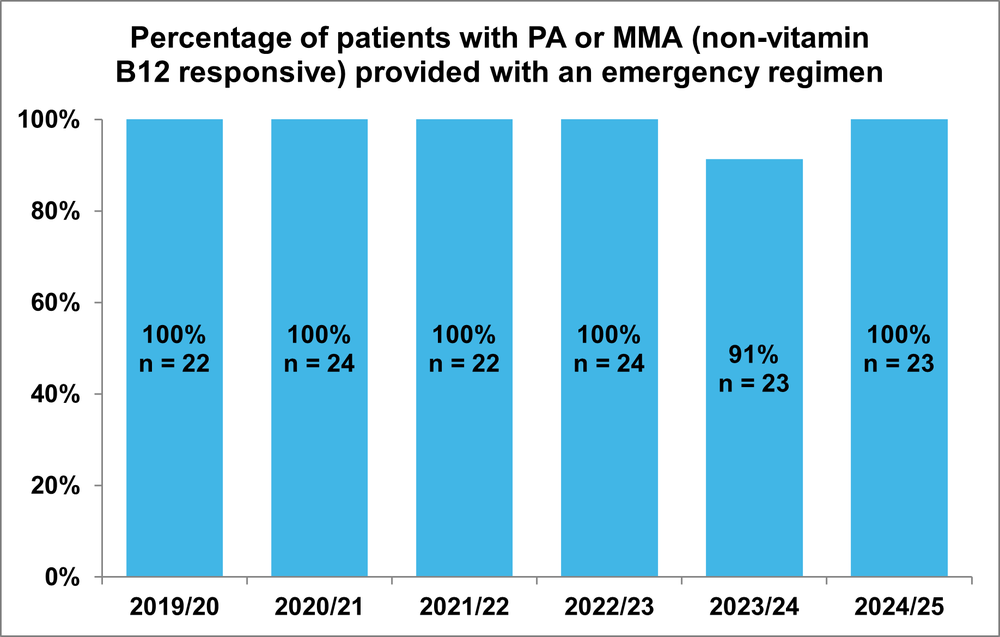
Figure 3.1 Percentage of patients with PA or MMA (non-vitamin B12 responsive) provided with an emergency regimen
These results show that consistently, 100% of our children and young people with these conditions are provided with an emergency regimen. This means that parents/carers are able to promptly implement the appropriate emergency regimen at home at the start of any illness that disrupts their child’s regular diet. This helps to avoid the co-occurrence of potentially serious metabolic problems when their child is unwell.
3.2 Patients with PA or MMA (non-vitamin B12 responsive) with an appropriate emergency regimen, reviewed at least yearly
To ensure that the emergency regimen is up-to-date for each patient, the regimen should also be reviewed at least yearly and adjusted accordingly.
This measure shows the number of children and young people with PA or MMA (non-vitamin B12 responsive) whose emergency regimen has been reviewed within 12 months. To ensure relevance of the data, patients who moved countries, transitioned to adult health services or died prior to their review date are not included in the figures.
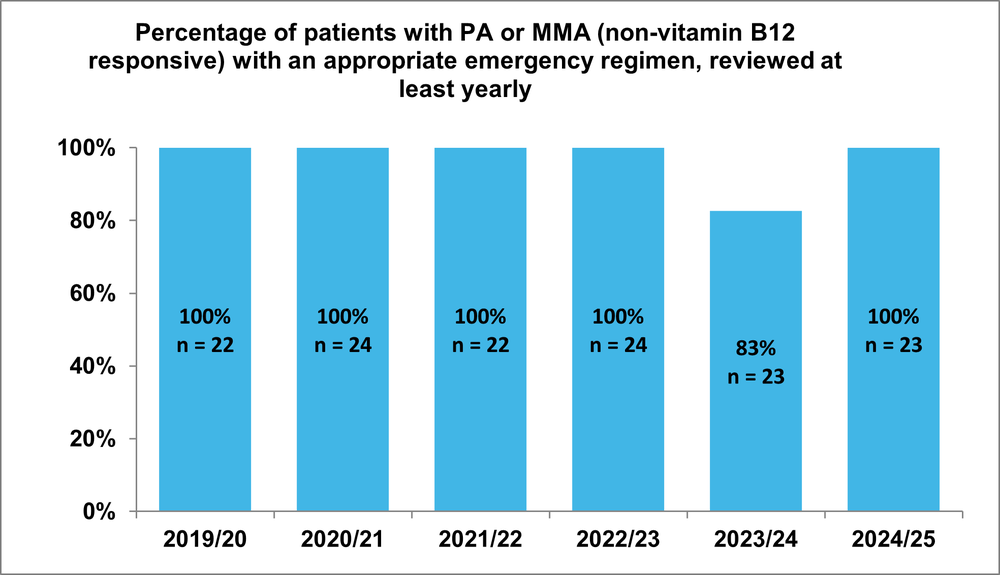
Figure 3.2 Percentage of patients with PA or MMA (non-vitamin B12 responsive) with an appropriate emergency regimen, reviewed at least yearly
Our results show that we have a near 100% achievement of yearly review of emergency regimens for our children and young people with PA or MMA (non-vitamin B12 responsive). This means that if they do become unwell, they can be given an up-to-date emergency regimen diet that is most appropriate for their age and weight to support their metabolic function during sickness.