https://www.gosh.nhs.uk/conditions-and-treatments/conditions-we-treat/pyruvate-dehydrogenase-deficiency/
Pyruvate dehydrogenase deficiency
Pyruvate dehydrogenase (PDH) deficiency is a rare inherited metabolic disorder that affects how the body can break down food and drink to make energy for every cell in the body. Pyruvate is a substance that plays an important part in the process to break down glucose. When PDH is deficient (too low), pyruvate is converted into lactic acid, which then builds up in the body. This information sheet from Great Ormond Street Hospital (GOSH), produced in association with the Freya Foundation, describes the symptoms and causes of PDH deficiency and how it can be treated.
Pyruvate dehydrogenase (PDH)
Our bodies are like living machines. Just like a car, we need fuel to work. The fuel for our bodies is the food that we eat. The body breaks down the food we eat to take out the goodness (nutrients) and turns it into energy. Every cell in the body needs energy so it can work properly.
The way our body breaks down food into energy is called metabolism. This is a series of complicated chemical reactions. Each nutrient needs a different chemical reaction to turn it into energy. Enzymes are the substances that trigger or catalyse the chemical reaction.
Pyruvate dehydrogenase (PDH) is an enzyme needed to convert the carbohydrates you eat into energy. Specifically, PDH turns a chemical called pyruvate into another chemical called acetyl CoA.
In PDH deficiency, the enzyme is less active than normal. This leads to an inefficiency in making energy, especially from carbohydrates (sugary and starchy foods such as cakes and sweets, and bread, pasta and rice). Also, instead of turning into acetyl CoA, the pyruvate is converted into a chemical called lactic acid.
This means there is a build-up of lactic acid and there is not enough energy to supply the body. This lack of energy especially affects the brain, as it needs a lot of energy. That is the reason why many of the symptoms of PDH deficiency are neurological (affecting the brain, spine or nerves, such as seizures and poor coordination).
PDH deficiency is a very rare condition, affecting fewer than 1 in 50,000 people.
What causes pyruvate dehydrogenase deficiency?
Pyruvate dehydrogenase deficiency is a genetic condition, caused by a mutation (change) in specific genes. Research has identified the most commonly affected gene as the PDHA1 gene – this affects 80 per cent of people with the condition. Other genes have also been identified as causing pyruvate dehydrogenase deficiency – the PDHB, DLAT, PDHX, DLD, and PDP1 genes – but these are less common.
All these genes are responsible for making parts of the PDH enzyme that is involved in breaking down carbohydrates into energy to be used by the body. As the PDH enzyme cannot be made properly, the body cannot convert the food and drink so there is not enough energy to function.
The gene mutation can be passed on from parent to child but in most cases with PDHA1 mutations, the mutation arises sporadically (out of the blue).
In cases where a mutation in the PDHA1 gene is inherited, it is passed on in an X-linked manner – that is, the mutation is on the sex chromosome (X-chromosome). Females have two X-chromosomes but in most cases one of them is usually dormant (inactive) and therefore females carrying one faulty copy and one normal copy of the gene can either be affected with symptoms of PDH deficiency or completely healthy, depending on whether the normal copy of the gene is active or not. Males only have one X-chromosome (and one Y-chromosome) so cannot compensate for the faulty one. This means that the most common form of PDH deficiency affects males.
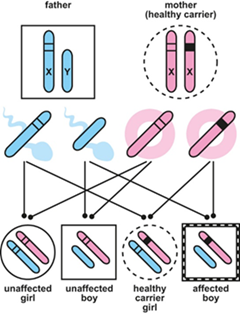
X-linked inheritance- how boys can inherit the affected gene
If the mutation affects the other genes, it is on one of the autosomes, that is, not the sex chromosomes. It is passed on in an autosomal recessive manner – this means that a child has to inherit the faulty gene from both parents to develop the condition. Although they have the faulty gene, parents do not usually show any symptoms of pyruvate dehydrogenase deficiency.
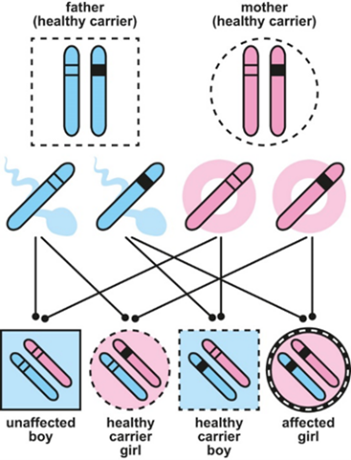
X-linked inheritance- how girls can inherit the affected gene
What are the symptoms of pyruvate dehydrogenase deficiency?
PDH affects each child differently although common symptoms include:
- developmental delay (this means that children do not reach key milestones in physical, emotional, social, communication, thinking, and learning skills as expected)
- fatigue/lack of energy
- seizures
- weak muscles
- poor coordination
- poor feeding
- poor motor skills (such as sitting, walking)
- nausea and vomiting
- poor growth
- breathing problems
How is pyruvate dehydrogenase deficiency diagnosed?
PDH deficiency may be suspected if a child has raised lactic acid levels or any of the symptoms described above.
Tests which may be done to confirm the diagnosis:
- Blood test to measure lactic acid and pyruvate levels
- Lumbar puncture to measure lactic acid and pyruvate levels in the fluid surrounding the brain and spinal cord (cerebrospinal fluid, CSF)
- MRI scan to look at the structure of the brain
- Skin biopsy to grow cells to measure the activity of the PDH enzyme
- Genetic testing (looking for changes in someone’s genes)
Information about these tests is available on our website at www.gosh.nhs.uk
A prenatal (before birth) diagnosis can be performed by analysing a known familial mutation (an inherited change passed down from a biological parent) in a fetus after biopsy (a small sample of tissue taken to be examined under a microscope and tested chemically in the laboratories) of chorionic villi (a type of tissue in the placenta) or amniocentesis (cells from the fluid surrounding the baby in the womb) depending on the gestational age (how far along the pregnancy is).
How can pyruvate dehydrogenase deficiency be treated?
Treatment aims to manage symptoms, as unfortunately there is no cure for PDH deficiency. Treatments which may help include:
- Anti-epileptic medicines can be used to treat seizures
- A ketogenic diet
A ketogenic diet is a special diet that is high in fat and low in carbohydrate.
It is started in hospital with advice and monitoring from a specialist dietitian, who will continue to advise you when you are back at home.
A low carbohydrate diet will stop the build-up of lactic acid.
A high fat diet will increase the production of ketones, which are an alternative source of energy for the brain.
- Dietary supplements
Your child may be prescribed some dietary supplements:
- Thiamine is a vitamin that helps the PDH enzyme to work more efficiently.
- Coenzyme Q10 is a supplement that helps our cells to make energy.
What is the outlook for children and young people with pyruvate dehydrogenase deficiency?
We know that this is a sensitive issue to talk about, but recognise it is a very important question for families.
The outlook is variable depending on the age at which symptoms appeared and the severity of symptoms. Unfortunately, babies who show symptoms soon after birth may not survive beyond a year.
Children who survive the first year will have severe learning disabilities. Those who show symptoms later in childhood do better because they can process a certain amount of nutrients, but they will still have learning disabilities.
Further information and support
Visit the GOSH website for more information about the Metabolic Medicine team at GOSH:
Metabolic Medicine team at GOSH
The Freya Foundation offers support and advice to anyone affected by pyruvate dehydrogenase deficiency. Visit their website at www.thefreyafoundation.co.uk or email them at thefreyafoundation@gmail.com
Metabolic Support UK (previously known as CLIMB) is the main umbrella organisation the UK offering advice and support to anyone affected by a metabolic condition. Call their helpline on 0800 652 3181 or visit their website at Metabolic Support UK